![]()
![]()
|
Bottomline: Myotonic dystrophy (DM1) is a genetic condition that affects many parts of the body e.g. muscles, heart, eyes, bowels and brain. It is autosomal dominant meaning that an individual needs to only inherit one abnormal gene change from one parent to be at risk for DM1. When symptoms start and how serious those symptoms are varies from person to person even within the same family. There are two kinds of genetic test results: normal (negative), premutation and positive (expanded). |
This resource is intended to facilitate discussion between an individual and their healthcare provider about genetic testing for DM1.
For support when genetic test results are reported see the GECKO on the run Myotonic Dystrophy Type 1: Genetic test results.
What is DM1?
DM1 is a multi-system condition and the main symptoms include:
- Myotonia: The muscles are not able to relax after an individual contracts their muscle. Patients experience stiffness or locking of the hands, sometimes also other muscles (jaws, tongue, legs)
- Slowly progressive muscle loss and muscle weakness, usually affecting the muscles of the feet, hands, and face first with gradual involvement of other muscles. This may lead to walking, swallowing, speech, and/or breathing difficulties. Loss of strength in the hand is frequently reported as an inability to open jars.
- Cataracts (clouding of the lens of the eye), usually at a younger than expected age.
- Conduction defects and arrhythmias (impaired electrical system of the heart and irregular heart rhythms).
- Fatigue and daytime sleepiness, disrupted sleep.
- Chronic diarrhea and/or constipation (often misdiagnosed as irritable bowel)
Other conditions or features that may be present: Type 2 diabetes, underactive thyroid, reduced fertility, and hair loss. Not everyone with DM1 will develop the same symptoms or all the symptoms or develop symptoms at the same age. This is true even among members of the same family.
Individuals with a genetic and/or clinical diagnosis of DM1 are monitored for these conditions. Treatment focuses on managing symptoms, preventing worsening or life-threatening complications and supporting overall health. There is no cure for DM1 yet, but this is an active area of research.
A Genetics Minute
Inside every cell of our body- muscle cells, brain cells – there is DNA. DNA is like a library of recipes that our bodies use to make important proteins - molecules that carry out essential jobs for development, maintenance, and health. These ‘recipes’ are called genes.
There are about 21,000 genes in the DNA library. The genes are arranged like beads on a string into structures called chromosomes. All of the genes come in pairs and one set is inherited from the egg (e.g. mother) and one set is inherited from the sperm (e.g. father). The DNA code is made up of small building blocks called adenine, cytosine, thymine, and guanine (A, C, T, G for short). These letters form the instructions for making the proteins.
Most (99.9%) of the DNA library is the same from person to person, but still this small bit of difference makes each of us unique. Sometimes a difference in our DNA does not have any effect on our health/development, but sometimes a difference as small as a switch between DNA building blocks (e.g. a change from an A to a C) can have a significant effect on health/development. These harmful gene changes are sometimes called pathogenic variants or mutations or abnormal.
What causes DM1?
DM1 is caused by a change in a gene called DMPK. In the DMPK gene, there is a section of DNA with the three-letter code 'CTG’ - representing the DNA base pairs cytosine, thymine, and guanine- which is repeated over and over again.
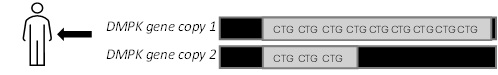
If an individual has too many CTG repeats (i.e. more than 50) on one of their two copies of the DMPK gene (a repeat expansion), they are more likely to develop symptoms. Symptoms tend to occur earlier in life and be more severe with a higher number of CTG repeats. DM1 is an autosomal dominant inherited condition meaning an individual needs to only inherit one abnormal gene from one parent to be at risk. This means their siblings and children have a 50% chance to inherit DM1 as well. When an abnormal DM1 gene is passed from parent to child there is a risk for the number of CTG repeats to increase and there is a chance that an even mildly affected parent can have severely affected child (e.g. congenital DM1).
Most laboratories cannot accurately 'count' the CTG repeats beyond a certain number. Because of this, individuals often receive either a negative or positive result only, and not their repeat number. Knowing the repeat number can be important for: understanding how significant an individual's symptoms could be, family planning, or determining clinical trial eligibility.
Who should be considered for DM1 genetic testing?
Consider offering genetic testing if an individual has:
- Been assessed by a neurologist or neuromuscular specialist or other specialist and there is clinical suspicion of DM1.
- Those with minor or few DM1 symptoms and an affected first-degree relative. (Assessment by specialist is still recommended prior to consideration of genetic testing)
- Had previous genetic testing for DM1 that was positive, but the CTG repeat number was not reported.
For individuals who have a family history of DM1 but no clinical symptoms (asymptomatic), they should be offered genetic counselling prior to genetic testing. This type of testing is called predictive testing. You can refer to your local genetics centre (www.geneticseducation.ca > Find your local genetics expert > Clinics) or obtain genetic counselling from a board-certified genetic counsellor through a private service. You can search through the Canadian Association of Genetic Counsellors directory at https://www.cagc-accg.ca/ > Find a Clinic.
What are the possible genetic test results?
The genetic testing result reports the number of CTG repeats (or repeat expansions) detected on both copies of the DMPK gene. There will be two expansion numbers reported as there are two copies of the gene, one inherited from each parent. The number of repeats is interpreted as:
- Normal (less than or equal to 49 CTG repeats). If both repeat expansions are within this range, an individual is not at increased risk for DM1. If there was clinical suspicion of DM1, this diagnosis is unlikely.
- Premutation (35-49 CTG repeats). If one repeat expansion is within this range (and the other normal), an individual is not at increased risk for DM1. If there was clinical suspicion of DM1, this diagnosis is unlikely. However, this individual’s children have a higher chance of developing DM1 because the CTG repeat can increase in size when passed on from parent to child.
- Positive (expanded, greater than or equal to 50 CTG repeats). If one repeat expansion is within this range (and the other normal), an individual is at increased risk for symptoms of DM1 and will develop symptoms of DM1 during their lifetime, or this confirms a clinical diagnosis of DM1. This individual’s siblings and children have a 50% chance to inherit DM1. The number of CTG repeats cannot predict the precise age of when symptoms start or how serious they will be. Results are generally interpreted as:
- 50-~150 repeats can cause mild DM1. Individuals may only have myotonia (muscles that do not relax as they should), cataracts, cardiac arrythmia and type 2 diabetes. Age of onset can vary among adults. Generally, these patients have fewer symptoms and get them later in life
- ~100-1000 repeats can cause classic DM1. Symptoms usually start in early adulthood and are more severe than mild DM1.
- Greater than 1000 repeats is expected to cause congenital DM1. This is when symptoms are present before or at birth, are severe, associated with learning difficulties and can be life-limiting.
Other result types (e.g. both positive) would be rare unless there is a family history consistent with DM1 on both sides of the family. Genetic counselling would be recommended.
Are there benefits to genetic testing for DM1?
Genetic testing for DM1 can:
- Provide an explanation for an individual’s symptoms with symptomatic treatments available.
- End a diagnostic odyssey with unnecessary tests and potentially harmful or ineffective treatments.
- Allow for earlier monitoring of symptoms and prevention of potentially life-threatening events when testing unaffected relatives, if positive.
- Help individuals and their healthcare practitioners understand the risk of DM1 to relatives.
- Provide information to help with reproductive decision making for those who are planning their families e.g. understanding the risk for congenital DM1.
- Determine eligibility for research/clinical trials (exact CTG repeat number may be needed).
- Connect with advocacy groups and the DM community for support and education.
Are there limitations or additional considerations to genetic testing for DM1?
- Positive results may cause or increase anxiety, depression, or other negative feelings.
- The number of CTG repeats cannot predict the precise age of when symptoms will present or how serious the symptoms could be.
- Risk of genetic discrimination: A positive genetic test result could lead to a diagnosis of DM1. This could impact an individual and their relatives’ insurance applications (e.g. life, disability, travel). There is a law in Canada called the Genetic Non-Discrimination Act (GNA) which protects individuals from having to share genetic results with third parties like employers and insurance companies. However, insurers can still request information if an individual or their relative has symptoms and/or a diagnosis. Insurers can also request non-genetic testing that may have been ordered after a positive genetic result (e.g. cardiac testing) and/or ask for family history.
How is the genetic test ordered?
If genetic testing is accepted by an individual, their healthcare practitioner can complete the Myotonic Dystrophy Canada/Children’s Hospital of Eastern (CHEO) genetic test requisition. The individual can then take this paperwork and go to a community blood laboratory for the blood draw. Their sample will be shipped to CHEO for analysis. Results can take 1-2 months.
If there is difficulty finding a blood draw location, please contact Muscular Dystrophy Canada at https://www.muscle.ca/.
What are the next steps?
All results will be reported to the healthcare practitioner that ordered the test by fax. The individual who had testing will receive a phone call from a CHEO laboratory genetic counsellor about their test result. This is an information session and not intended to replace a genetic counselling session. A referral to the local genetics centre may still be considered and/or indicated.
See our Post-test Point of Care tool for how to proceed once results are available.
Resources
- Muscular Dystrophy Canada: https://muscle.ca/
- Myotonic Dystrophy Foundation: https://www.myotonic.org/
- Genetics Education Canada: Knowledge Organization https://www.geneticseducation.ca > Resources for Clinicians >Neuromuscular
This resource was developed by Genetics Education Canada: Knowledge Organization with support from Muscular Dystrophy Canada.

