
Download the comprehensive GECKO Messenger, the quick reference GECKO on the run, and/or the point of care triage tool.
Under review. Last updated January 2019
Bottom line: Lynch syndrome (LS), also known as Hereditary Non-Polyposis Colorectal Cancer (HNPCC), is the most common hereditary colorectal cancer predisposition syndrome. It is an autosomal dominant condition that results in an increased lifetime risk of colorectal cancer (CRC) in addition to other cancers. Individuals at high or intermediate risk of LS should be referred for a genetic consultation for consideration of genetic testing. The results of genetic testing can lead to appropriate surveillance and improved outcomes for both affected individuals and their family members. Conversations between patients and their healthcare providers are strong drivers of screening participation.
What is Lynch Syndrome?
It is estimated that one in 14 men and one in 15 women will develop CRC during their lifetime. CRC is the second deadliest form of cancer, but, with early detection, there is a 90% chance of cure1,2. About 5-10% of colorectal cancer is hereditary.
Lynch syndrome (LS), also known as Hereditary Non-Polyposis Colorectal Cancer (HNPCC), is the most common inherited cause of CRC3. LS accounts for about 0.7-3.6% of cases of CRC4. Research on LS-related endometrial cancer is still emerging; current data suggest that in North America between 1.8% and 4.5% of cases are attributed to LS4. Having an LS mutation results in an increased lifetime risk of CRC and other Lynch syndrome –related cancers (see What do the results mean?)
Other hereditary CRCs, such as Familial Adenomatous Polyposis [FAP] account for about 1% of CRC. This GEC-KO Messenger will focus on LS and will not deal further with FAP.
What do I need to know about the genetics of Lynch Syndrome?
LS is caused by an inherited mutation in one of four mismatch repair (MMR) genes (MLH1, MSH2, MSH6, PMS2) or in EPCAM, a gene that plays a role in MSH6 gene inactivation. Mismatch repair genes play an important role in a cell’s ability to repair DNA damage that occurs as a cell grows and divides, by identifying and removing single nucleotide mismatches, insertions and deletion loops. Defects in the MMR pathway lead to an accumulation of mutations in a cell which may result in a malignancy3.
Microsatellite Instability (MSI)
LS is characterized by tumours that exhibit microsatellite instability (MSI). A microsatellite is an area of DNA with a repetitive sequence (i.e. CGCGCGCGC or GAAGAAGAA). These stretches of DNA are susceptible to changes in the number of repeats when a mutation in a MMR gene is present. Cancer arising as the result of a defective MMR gene exhibits an inconsistent number of microsatellite repeats when compared to normal tissue – this is called microsatellite instability (MSI). Approximately 90% of CRCs occurring in individuals with Lynch syndrome exhibit MSI. Approximately 15% of sporadic colorectal cancers (not associated with LS) also exhibit MSI.5
Pattern of Inheritance
LS is an autosomal dominant condition with reduced penetrance and variable expressivity. This means that not all individuals who inherit a mutation in an LS gene will develop cancer (reduced penetrance) and the signs and symptoms/type and onset of cancer will vary between affected family members (variable expressivity).
Who should be offered genetic testing?
Currently the decision to have genetic testing is made in the setting of a genetics consult at a hereditary cancer program or a general genetics clinic. Click to connect to your local genetics centre or hereditary cancer program.
If possible, the affected individual in the family at highest risk to carry a mutation is offered testing first in order to maximize the likelihood of detecting a mutation. This would usually be a young individual with CRC or another LS-associated cancer.
How do I assess my patient’s risk?
LS can be identified by looking for red flags in a personal and/or family history.

Personal history Red Flags for a CRC syndrome6:
These are general triage guidelines to identify patients at high risk for LS. You should check with your local genetics centre or hereditary cancer program for more specific details.
Consider referring your patient if he/she has
An early age of CRC diagnosis (<50 years)*. Diagnosis under 35years is more likely to be LS.
An early age of endometrial cancer diagnosis (<50 years)
Multiple primary LS-related cancer diagnoses, regardless of age*
A CRC diagnosis and one or more 1st degree relatives with a LS-related cancer, with one of the cancers being diagnosed <50 years*
A CRC diagnosis and two or more 1st or 2nd degree relatives with LS- related cancers regardless of age <50 years*
A CRC diagnosis <60 years with histological features suspicious for LS*(excess infiltrating lymphocytes, mucinous/signet cell features, Crohn’s-like reaction), particularly when primary tumour is right sided
The Bethesda criteria are those marked with an asterisk*. They were developed to identify people at risk of having a LS gene mutation who do not have a strong family history of LS-related cancers. Amsterdam criteria are used to identify individuals at risk of carrying a LS gene mutation based on a strong family history of LS-related cancers. Patients who meet either Bethesda or Amsterdam criteria are at high risk for LS. Please refer to the Genetics Education Canada- Knowledge Organization (GEC-KO) glossary for more information about Bethesda and Amsterdam criteria.
Family history Red Flags for a CRC syndrome:
Take a three generation family history (your patient’s generation and, depending on your patient’s age, a prior generation and a subsequent generation, or two prior generations including both maternal and paternal sides of the family); target cancer (all types) and polyps, noting the ages at diagnosis and the primary tumour sites.
You should consider referring your patient to your local genetics centre or hereditary cancer program for further assessment if they are at high risk for hereditary CRC syndrome.
A patient is considered to be at high risk for LS syndrome if they:
Has a known LS causing mutation in the family
If they meet the revised Amsterdam criteria6 , meaning they they have at least three relatives with a cancer associated with LS (Box 1); the following criteria should also be present:
- One must be a first degree relative of the other two;
- At least two successive generations must be affected (autosomal dominant inheritance);
- At least one relative with LS-related cancer should be diagnosed before age 50
Tumour pathology should be verified when possible and other CRC syndromes should be ruled out
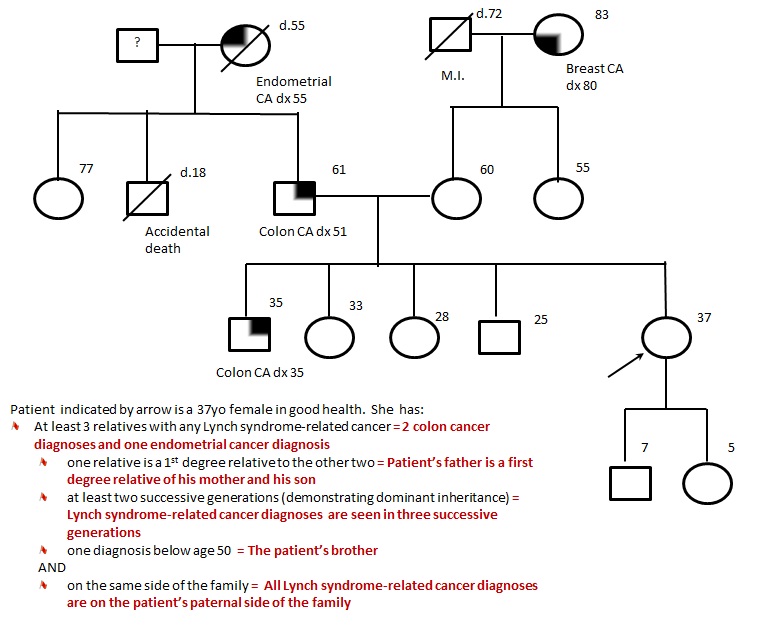
Figure 1. Example of a pedigree meeting Amsterdam criteria. The patient (37yo female indicated by the arrow) could benefit from genetic counselling based on her family history; however she would likely not be offered genetic testing because an affected family member (ideally her affected brother) would be most appropriate and informative to test first.
If your patient does not meet any of the criteria above, but you are suspicious of a hereditary cancer syndrome, consult your local genetics centre or hereditary cancer program. Please refer to hereditary cancer syndrome point of care tool for further information.
How do I order the genetic test?
Currently the decision to offer genetic testing is made in the setting of a genetics consult at a hereditary cancer program or general genetics clinic. To assess if your patient could be eligible for genetic testing see Who Should Be Offered Genetic Testing? Click to connect with your local genetics centre or hereditary cancer program and find their referral criteria. If your patient does not have cancer, genetic testing of a relative with cancer will be recommended as a first step
Once a patient has been referred to a hereditary cancer program/general genetics clinic and genetic testing is offered and accepted, the algorithm for LS testing (Figure 2) is as follows. Ideally testing begins with immunohistochemical (IHC) analysis of a CRC tumour (it may be possible to test other tumour types if a CRC tumour is not available) for the proteins associated with the LS genes, MLH1, MSH2, MSH6, and PMS2. IHC analysis looks at the protein products of the LS genes. Inherited mutations in MLH1 result in loss of expression of MLH1 and PMS2 and inherited mutations in MSH2 typically result in loss of expression of MSH2 and MSH6. If IHC analysis reveals a protein to be deficient, genetic testing can be offered to the affected individual and performed on a blood sample. If IHC analysis does not clearly show protein deficiency, the next step is microsatellite instability (MSI) testing of the tumour sample. If MSI is stable or low, no further testing is indicated. If MSI is high, genetic testing can be offered to the affected individual and performed on a blood sample. Some centres will arrange IHC or MSI alone, others will carry out both tests at the same time.
Figure 2. Lynch syndrome testing algorithm.
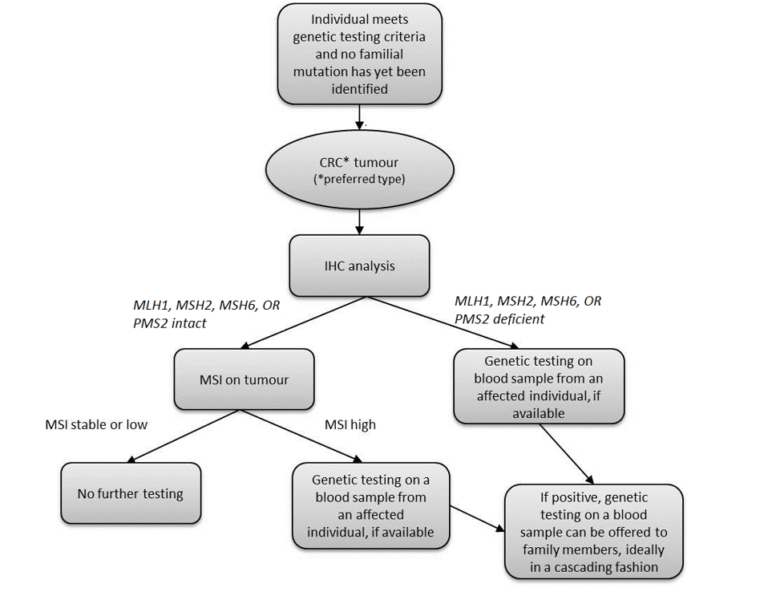
*Individuals who meet Amsterdam criteria may be able to pursue genetic testing if there is no CRC or LS-related tumour to test.
What do the genetic test results mean?
If your patient has been found to carry a mutation in a Lynch syndrome gene, he/she has an increased lifetime risk to develop certain cancers (Table 1)3. This also means that family members are at risk of carrying the same mutation and of having similar cancer risks. Evidence is emerging from population based studies that these cancer risks are gene specific8,9.
Table 1. Lifetime extra-colonic cancer risks for individuals who have inherited a mutation in a LS gene as compared to the general population.6

Table 2: Lifetime extra-colonic cancer risks for individuals who have inherited a mutation in a LS gene as compared to the general population.6
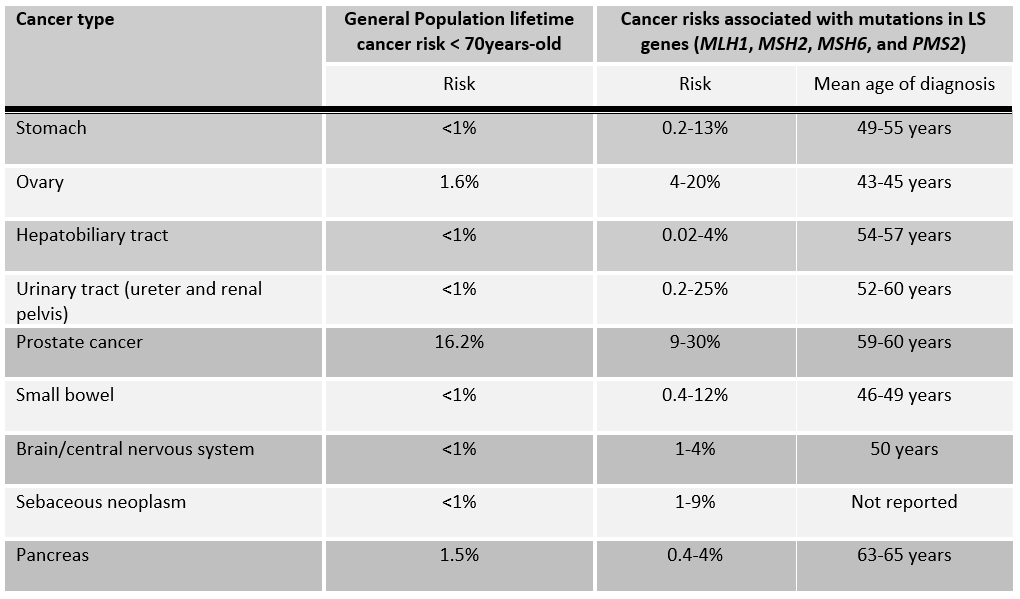
*The cancer risks cited in Tables 1 and 2 are associated with mutations in MLH1, MSH2, MSH6, and PMS2, and do not include cancer risks associated with mutations in EPCAM as these associated risks are not well characterized.
How will genetic testing help you and your patient?
If a mutation is identified (a positive test result):
- Appropriate surveillance and management can improve outcome
- High compliance with screening leads to no increase in mortality for individuals with Lynch syndrome over their mutation-negative relatives3,9
- When detected early, colorectal cancer has a 90% cure rate1
- Studies show that conversations between patients and providers are the strongest driver of screening participation1,2
- Other at risk family members can be identified10
- Positive health behaviours can be reinforced10
If a mutation is not identified and testing was for a known familial mutation (true negative):
- Your patient is not considered to be at increased risk of developing hereditary cancer but may still be at increased risk of cancer depending on family history
- You can provide reassurance to your patient
Are there harms or limitations of genetic testing?
If a mutation is identified (a positive test result)10:
- Your patient may experience psychological distress knowing he/she is at increased risk to develop cancer, and/or over the possibility he/she may have passed the mutation to his/her children
- Family issues such as confidentiality concerns may inhibit the transfer of information between relatives
- Incomplete penetrance – being identified as having a mutation does not mean one will develop cancer
- Historically, genetic testing results may have affected an individual’s ability to obtain life, disability, critical illness, long-term care and/or extended health insurance. However, in 2017 Canada passed the Genetic Information Non-Discrimination Act (GNA) that protects individuals from having their genetic test results used to prevent them from obtaining insurance.
If a mutation is not identified in an unaffected patient and testing was for a known familial mutation (true negative)10,11,12:
- Depending on family dynamics (some siblings may have tested positive while some have not) or family history of extensive cancer, your patient may experience psychological distress such as ‘survivor guilt’ or ‘identity loss’
- Your patient may develop a complacent attitude to health and screening
If no mutation is identified in an affected patient who has no known familial mutation (uninformative result) or when a variant of uncertain significance (VUS) is identified10,12:
- The diagnosis of Lynch Syndrome is not confirmed or ruled out, even in families with a strong history of CRC
- Screening recommendations will be based on a combination of factors, such as family history and in cases where a VUS was identified, information about the VUS
- Please consult your patient’s geneticist, also see surveillance and management
Where do I refer my patient?
Click to connect to your local genetics centre or hereditary cancer program.
Note that hereditary cancer programs/general genetics centres vary with regards to the referrals they choose to accept. You may want to contact your local genetics centre or hereditary cancer program for more information.
Include all relevant information with your referral (e.g. family history, cancer history, pathology results, genetic test results, and results of investigations such as colonoscopies). Encourage your patient to collect this information if you do not have it, to facilitate a productive genetic counselling session and to prevent unnecessary delays when further clarification is needed before an appointment can be booked.
Surveillance and Management
High risk for CRC (carriers of a Lynch syndrome gene mutation and their first degree relatives who have not yet had genetic testing):
Colorectal Cancer
Current recommendations for those with LS mutations are colonoscopy every 1-2 years beginning between ages 20 and 25, or 2-5 years prior to the earliest colon cancer in the family if that diagnosis was made before age 25 years, whichever is earlier.3,5,12 Recommendations may vary depending on the specific mutation.3,10 Because routine colonoscopy is an effective preventive measure for colorectal cancer, prophylactic colectomy is generally not recommended for individuals with LS.3
Endometrial and Ovarian cancer
Screening for endometrial or ovarian cancer may include transvaginal ultrasound and endometrial biopsy, however, there is little evidence of the effectiveness of these tests.12 Most importantly, people should be educated about the symptoms of endometrial cancer because many endometrial cancers can be diagnosed at early stages on the basis of symptoms.3,10 Prophylactic hysterectomy and bilateral salpingo-oophorectomy is a risk-reducing option that individuals with LS who have completed childbearing can consider.3,5,12
Other Extracolonic cancers5
There is no clear evidence to support additional screening for gastric, duodenal, small bowel, central nervous system or breast cancer. Annual health examination is recommended.
However, for some individuals, depending on family history or ancestry (Asian), upper endoscopy surveillance with extended duodenoscopy can be used to screen for cancer of the stomach every 3-5 years beginning at age 40.
Screening for urothelial cancer by urinalysis starting at age 30-35 years may also be considered, if there is a family history of these cancers. At this time, there is no clear evidence to recommend any particular screening strategy for these cancers.
Individuals who have tested negative for a known familial LS gene should follow provincial guidelines for population risk CRC screening (see below). For those individuals who have a family history of CRC unrelated to the mutation in their family (i.e. on the other side of the family), screening recommendations will be based on that family history. Consult your local genetics centre or hereditary cancer program.
For individuals where no mutation was identified and there was no known familial mutation (uninformative result) or when a variant of uncertain significance (VUS) was identified, screening recommendations will be based on a combination of factors, such as family history and in cases where a VUS was identified, information about the VUS.
Increased risk for CRC13
Table 3. Screening recommendations for individuals at increased risk to develop CRC and who do not meet high risk, LS criteria.
| Criteria | Recommendation |
| One or more 1st degree relatives with a CRC diagnosis at any age | Colonoscopy beginning at age 40 OR 10 years younger than the youngest CRC diagnosis. Repeat every 3-5y depending on family history and findings. |
| One or more 2nd degree relatives with a CRC diagnosis less than age 50 | Colonoscopy beginning at age 50. Repeat every 5-10y or as indicated by positive findings. |
| A 1st degree relative with advanced adenomas | Colonoscopy beginning at age 40 OR at age of detection in relative, whichever is first. Repeat every 5-10y or as indicated by positive findings. |
| A personal history of colorectal adenomatous polyps
|
Colonoscopy repeated as indicated by colonoscopy findings, management is dependent upon findings. |
| A personal history of inflammatory bowel disease
|
Initiate screening 8y after onset of symptoms (consult specialist), with colonoscopy every 1-3 years. Management is dependent upon findings. |
General Population Risk for CRC
For patients who are at general population risk for CRC, recommendations should follow provincial guidelines. In Ontario, these can be found at Cancer Care Ontario under the ColonCancer Check program. General population screening guidelines are for individuals who have no symptoms of CRC and no family history of CRC, or who test negative for a known LS gene mutation in the family.
For all average risk adults 50 years and older, recommended screening for CRC is Fecal Occult Blood Testing (FOBT) every two years.
Resources for health professionals
[1] Cancer Care Ontario (CCO). https://www.cancercare.on.ca/ [Accessed January 2019].
[2] ColonCancerCheck (CCC) http://www.health.gov.on.ca/en/pro/programs/coloncancercheck/ [Accessed January 2019].
[3] GeneReviews. Lynch Syndrome. http://www.ncbi.nlm.nih.gov/books/NBK1211/ Updated April 12th, 2018,
[Accessed January 2019].
[4] Egoavil C, Alenda C, Castillego A, Paya A, Peiro G, Sánchez-Heras A, et al. Prevalence of Lynch syndrome among patients with newly diagnosed endometrial cancers. PLoS One. 2013; 8(11):e79737.
[5] Referenced with permission from the NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®) for Genetic/Familial High-Risk Assessment: Colorectal V.1.2018 © National Comprehensive Cancer Network, Inc 2018. All rights reserved. [Accessed January 31, 2019]. To view the most recent and complete version of the guideline, go online to www.nccn.org.
[6] Giardiello FM, Allen AI, Axilbund JE, Boland CR, Burke CA, Burt RW,et. al., Guidelines on genetic evaluation and management of Lynch Syndrome: A consensus statement by the U.S. Multi-Society Task Force on Colorectal Cancer. Gastroeneterology. 2014; 147(2): 502-526.
[7] Bonadona V, Bonaïti B, Olschwang S, Grandjouan S, Huiart L, Longy M, et al. Cancer risk association with germline mutations in MLH1, MSH2 and MSH6 genes in Lynch syndrome. JAMA. 2011; 305(22): 2304-2310.
[8] Dowty JG, Win AK, Buchanan DD, Lindor NM, Macrae FA, Clendenning M, et al. Cancer risks for MLH1 and MSH2 mutation carriers. Hum Mutat. 2012; 34(3): 490-497.
[9] Järvinen HJ, Renkonen-Sinisalo R, Akhtán-Collán K, Peltomäki P, Aaltonen LA, Mecklin JP. Ten years after mutation testing for Lynch syndrome: cancer incidence and outcome in mutation-positive and mutation-negative family members. J Clin Oncol. 2009; 27:4793-4797.
[10] Riley BD, Culver JO, Skrzynia C, Senter LA, Peters JA, Costalas JW, et al. Essential elements of genetic cancer risk assessment, counselling, and testing: Updated recommendations of the National Society of Genetic Counselors. J Genet Counsel. 2012; 21: 151-161.
[11] Valverde K. Why me? Why not me? J Genet Counsel. 2006; 15(6): 461-463
[12] Vasen HFA, Blanco I, Akhtan-Collan K, Gopie JP, Alonso A, Aretz S, et al. Revised guidelines for the clinical management of Lynch syndrome (HNPCC): recommendations by a group of European experts. Gut. 2013; 62:812–823.
[13] Referenced with permission from the NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®) for Colorectal Cancer Screening V.1.2018 © National Comprehensive Cancer Network, Inc 2018. All rights reserved. [Accessed January 31, 2019]. To view the most recent and complete version of the guideline, go online to www.nccn.org.
Additional resources for health professionals:
The Jackson Laboratory Risk Assessment and Screening Toolkit https://www.jax.org/education-and-learning/clinical-and-continuing-education/cancer-resources/crc-toolkit
The Jackson Laboratory, American Cancer Society and National Colorectal Cancer Roundtable have developed a toolkit for primary care providers to help them implement a structured family history collection system to identify and manage individuals at increased or high risk of colorectal cancer, and to facilitate timely diagnostic evaluation of patients with signs or symptoms of early onset colorectal cancer.
Resources for patients and the public
ColonCancerCheck (CCC) https://www.cancercareontario.ca/en/cancer-care-ontario/programs/screening-programs/colon-cancer-check-colorectal-screening
Canada’s first province-wide, population-based colorectal cancer screening program — ColonCancerCheck — launched in Ontario in 2008. The program is a partnership between the Ministry of Health and Long-Term Care and Cancer Care Ontario.
Canadian Cancer Society (CCS) http://www.cancer.ca/en/?region=on
Lynch Syndrome International (LSI) http://www.lynchcancers.com/
An all-volunteer organization founded and governed by LS survivors, their families, and health care professionals who specialize in LS. The primary mission is to focus on providing support for individuals affected by LS, creating public awareness of the syndrome, educating members of the general public and health care professionals and providing support for LS research endeavours.
Authors: S Morrison MS CGC, JE Allanson MD FRCPC, E Tomiak MD FRCPC, K Semotiuk MS (C)CGC and JC Carroll MD CCFP
Updated by the GECKO team: JC Carroll MD CCFP, JE Allanson MD FRCPC FCCMG, S Yusuf MS CGC
Disclaimer:
· GECKO is an independent not-for-profit program that does not accept support from commercial or non-academic entities.
· GECKO aims to aid the practicing non-genetics clinician by providing informed resources regarding genetic/genomic conditions, services and technologies that have been developed in a rigorous and evidence-based manner with periodic updating. The content on the GECKO site is for educational purposes only. No resource should be used as a substitute for clinical judgement. GECKO assumes no responsibility or liability resulting from the use of information contained herein.
· All clinicians using this site are encouraged to consult local genetics clinics, medical geneticists, or specialists for clarification of questions that arise relating to specific patient problems.
· All patients should seek the advice of their own physician or other qualified clinician regarding any medical questions or conditions.
· External links are selected and reviewed at the time a page is published. However, GECKO is not responsible for the content of external websites. The inclusion of a link to an external website from GECKO should not be understood to be an endorsement of that website or the site’s owners (or their products/services).
· We strive to provide accurate, timely, unbiased, and up-to-date information on this site, and make every attempt to ensure the integrity of the site. However, it is possible that the information contained here may contain inaccuracies or errors for which neither GECKO nor its funding agencies assume responsibility.

